A21 - A global role of alternative splicing in the secretory pathway: The COPII scaffold and beyond
Principal Investigator
Prof. Dr. Florian Heyd, FU Berlin
The transport of secretory proteins from the endoplasmatic reticulum (ER) to the Golgi depends on formation of the COPII protein scaffold at the membrane of ER exit sites (ERES). Assembly of the COPII coat enables cargo concentration and eventually budding of transport vesicles from the ER. Sec16 is a peripheral membrane protein localized to ERES that directs COPII scaffold assembly. It has been shown to be essential for ER-to-Golgi transport in cells, but its precise role remains elusive. Furthermore, it is largely unknown how COPII transport is regulated to accommodate tissue- or activation-specific differences in cargo load and identity. In our previous work, we have identified an alternative splicing switch in Sec16 to mediate increased efficiency of COPII transport upon T cell activation; this splicing switch allows T cells to cope with the increase of secretory cargo upon activation (Figure 1). As a mechanistic basis, we suggest that the C-terminal Sec16 domain represents a splicing-controlled protein-interaction platform, with the individual isoforms showing differential interactions with COPII components. This is the first evidence that the COPII pathway is modulated by alternative splicing, providing a new regulatory layer for protein secretion and its adaptation to changing cellular demands. In the current proposal, we will perform a comprehensive analysis of Sec16 splice isoforms and their role in COPII scaffold formation, from structures at atomic resolution to in vivo phenotypes in mouse models. We will use recombinant, purified Sec16 splice isoforms and investigate their interaction with components of the COPII machinery. We will then use these proteins to investigate the influence of Sec16 alternative splicing on COPII formation and vesicle budding in vitro. To perform such analyses in cells, we have used the CRISPR/Cas9 technology to generate cell lines with alternative Sec16 exons deleted. Such cell lines provide the unique opportunity to investigate the efficiency of COPII transport as a function of Sec16 isoform expression; in addition, these cell lines in combination with super-resolution microscopy will allow us to further analyze and understand the role of Sec16 in organizing ERES architecture. Finally, we aim to generate isoform-specific mouse models to investigate the role of Sec16 alternative splicing in controlling the early secretory pathway in vivo.
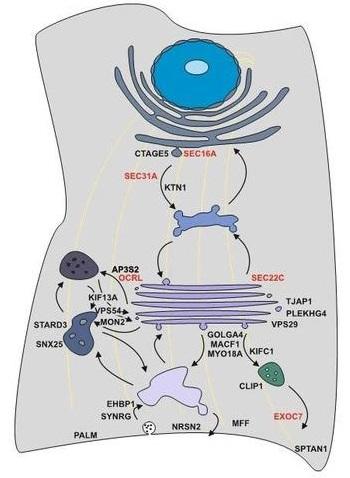
Figure 1: Model of splicing-regulated secretion modulators. Alternative splicing controls all steps of the secretory pathway. Selected candidates and their localization in the cell are shown. Alternative splicing of candidates in red has been confirmed to control protein secretion using the RUSH system.
References:
- Neumann A, Schindler M, Olofsson D, Wilhelmi I, Schürmann A, Heyd F. Genome-wide identification of alternative splicing events that regulate protein transport across the secretory pathway. J Cell Sci. 132(8):jcs230201 (2019)
-
Preußner, M., Wilhelmi, I., Schultz, A. S.., Finkernagel, F., Michel, M., Möröy, T. & Heyd, F. Rhythmic U2AF26 alternative splicing controls Period1 stability and the circadian clock in mice. Molecular Cell.54, 651-662 (2014)
-
Michel, M., Wilhelmi, I., Schultz, A. S., Preußner, M. & Heyd, F. Activation-induced Traf3 alternative splicing regulates the non-canonical NFkB pathway and chemokine expression in human T cells. Journal of Biological Chemistry289, 13651-13660 (2014)
-
Gaudreau, M. C., Heyd, F., Bastien, R., Wilhelm, B. & Möröy, T. Alternative splicing controlled by heterogeneous nuclear ribonucleoprotein L regulates development, proliferation, and migration of thymic pre-T cells. The Journal of Immunology188, 5377-5388 (2012)
-
Martinez, N. M., Pan, Q., Cole, B. S., Yarosh, C. A., Babcock, G. A., Heyd, F., Zhu, W., Ajith, S., Blencowe, B. J. & Lynch, K. W. Alternative splicing networks regulated by signaling in human T cells. RNA. 18, 1029-1040 (2012)
- Heyd, F. & Lynch, K. W. Degrade, move, regroup: signaling control of splicing proteins. Trends in Biochemical Sciences 36, 397-404 (2011) (Review)
-
Heyd, F. & Lynch, K. W. Phosphorylation-dependent regulation of PSF by GSK3 controls CD45 alternative splicing. Molecular Cell. 40, 126-137 (2010)
-
Möröy, T. & Heyd, F. The impact of alternative splicing in vivo: Mouse models show the way. RNA 13, 1155–1171 (2007) (Review)
-
Heyd, F., ten Dam, G. & Möröy, T. Auxiliary splice factor U2AF26 and transcription factor Gfi1 cooperate directly in regulating CD45 alternative splicing. Nature Immunology7, 859-867 (2006)